
Tadahiro Fujitani
Interdisciplinary Research Center for Catalytic Chemistry
National Institute of Advanced Industrial Science and Technology (AIST)

Yuta Hashiguchi
Corporate Research and Technology UBE Corporation
In recent years, the field of materials development has experienced a dramatic transformation: advances in computer performance and artificial intelligence have replaced the traditional approach to designing new materials—which relied heavily on experience and intuition, and which was highly inefficient—with the new paradigm of materials informatics, in which sophisticated information-processing algorithms, fueled by high-quality data, are exploited to accelerate the development of new materials. Inspired by this sea change, in 2016 we initiated the NEDO project Platform technology for ultra-rapid development of ultra-advanced materials, with the goal of achieving rapid development of functional materials by harnessing the "holy trinity" of computation, processing, and measurement. More specifically, taking organic functional materials as our model materials, we aimed to develop techniques for predicting the conditions under which specific material properties emerge; our framework for this was a data-driven research approach based on data created by the fusion of three core technologies1) (Figure 1): (1) innovative new computational technologies for predicting material properties using multiscale methods, (2) revolutionary new process technologies enabling actual sample prototypes to be fabricated rapidly at will, and (3) sophisticated new measurement technologies allowing precision measurements of structures that could not be measured via conventional metrology, as well as in-situ observation and discovery of novel functional properties. Our hypothesis was that, by developing this hybrid approach, we would reduce R&D times and establish techniques for discovering truly innovative new materials—materials that could not be found by simply extrapolating from existing materials into the future. To test this hypothesis, we assembled a research collaboration involving three institutions—Yokohama Rubber Company, UBE Corporation, and the Research Association of High-Throughput Design and Development for Advanced Functional Materials—to tackle two challenges: developing butadiene (BD) synthesis catalysts from ethanol (EtOH) and establishing flow-based methods for synthesizing Pd-core Pt-shell (Pd@Pt) catalysts.
In this article, we outline our computational approach, and describe our techniques for making the high-throughput of key experiments in catalyst chemistry to rapidly acquire a large amount of data—including automatic catalyst synthesis, rapid characterization of catalyst performance using multiple reactors, and rapid measurement of catalyst properties— and discuss our efforts to exploit data-driven catalyst informatics for automated catalyst discovery.
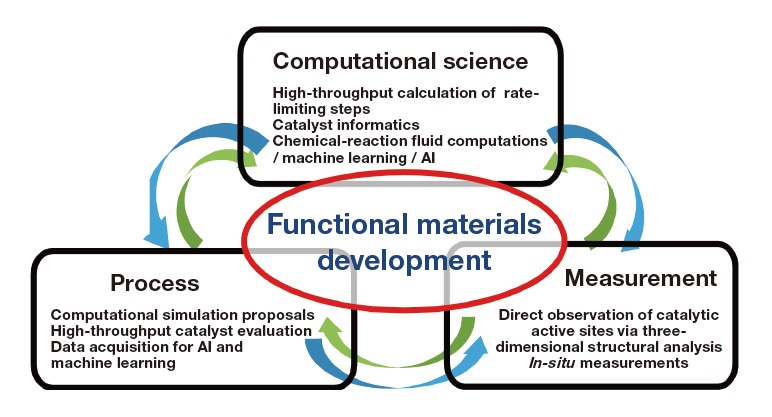
Fig. 1 Using a revolutionary new approach to material development to establish platform technologies for functional materials.
The reaction that converts EtOH to BD (the ETB reaction) is a complicated sequence of four processes: dehydrogenation, Aldol condensation, Meerwein-Ponndorf-Verley (MPV) reduction, and dehydration (Scheme 1) 2). This suggests that achieving optimal conversion efficiency and selectivity in the ETB reaction will require a catalyst containing an appropriate balance of acidic and basic reaction sites. However, exhaustive-search attempts to find the optimal catalyst for a given reaction are complicated by the large number of candidate catalysts that must be considered and the need to test catalytic activity under multiple reaction conditions—which, together, produce a combinatorial explosion in the number of individual tests that must be run, ensuring that the time and cost required for comprehensive screening quickly grow prohibitive. In this project we pursue an alternative approach: we introduce a collection of highthroughput laboratory instruments, gather copious data, and apply a data-driven approach based on catalyst informatics to develop new catalysts.
More specifically, in this study we use high-throughput instruments for three of the steps in our experimental procedure: preparing catalysts, evaluating catalytic activity, and characterizing physicochemical properties. For catalyst preparation, we used an automated catalyst synthesizer (Unchained Labs Big Kahuna, shown in Figure 2A) to prepare aqueous solutions of 50 varieties of metal-oxide precursors, which were then impregnated to an SiO2 support. This approach allowed the preparation of a total of around 200 catalysts, with varying loading amounts and calcination temperatures, in a relatively short period of time. We assessed catalytic activity using an 8-channel flow reactor (Taiyo System TS-14-8R, shown in Figure 2B) and a 16-channel flow reactor (Avantium Flowrence XR, shown in Figure 2C). Data were analyzed via online gas chromatography, and we obtained information on the composition of the reaction products produced by the EtOH conversion reaction. Dividing the ETB reaction into two stages, for the first stage we primarily considered the activity of the reaction converting EtOH into acetaldehyde (AcH), while for the second stage we primarily considered the activity of the reaction converting EtOH/AcH mixtures into BD.
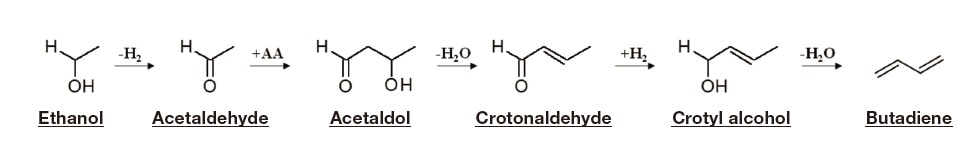
Scheme 1 Reaction mechanism for the ETB reaction that converts ethanol (EtOH) to butadiene (BD).

Fig. 2 High-throughput laboratory instruments.
A) Automated catalyst synthesizer. B,C) 8-channel and 16-channel flow reactors used to evaluate catalytic activity.
We first performed screening to identify catalysts that effectively catalyze the first-stage reaction converting EtOH to AcH. For this screening we evaluated a total of 200 candidate catalysts; Table 1 lists results for a representative subset of 10 of these candidates. We observe high rates of EtOH→AcH conversion for three catalysts i n particular—SiO2- supported CuO, Ag, and SnO2—indicating that these three candidates effectively catalyze the first-stage reaction3). For the ZnO catalyst, we observe conversion to BD as well as conversion to AcH. On the other hand, six of the candidate catalysts—IrO2, MoO3, Ga2O3, V2O5, Nb2O5, and NiO—have the effect of producing ethylene from EtOH, with EtOH dehydration constituting the primary reaction.
Having identified CuO, Ag, SnO2, and ZnO as effective catalysts for EtOH conversion to AcH4,5), we next investigate how the catalytic activity of these candidates is affected by reaction temperature. For the CuO catalyst, catalytic activity is high at the relatively low temperature of 623 K, but falls off dramatically as the reaction temperature is increased. For the Ag, SnO2, and ZnO catalysts, the AcH yield increased at higher reaction temperatures. In particular, the Ag/SiO2 catalyst exhibited the highest AcH selectivity and yield over a wide range of reaction temperatures.
Table 1 EtOH reaction activity for a subset of the catalyst candidates prepared by the high-throughput catalyst synthesizer.
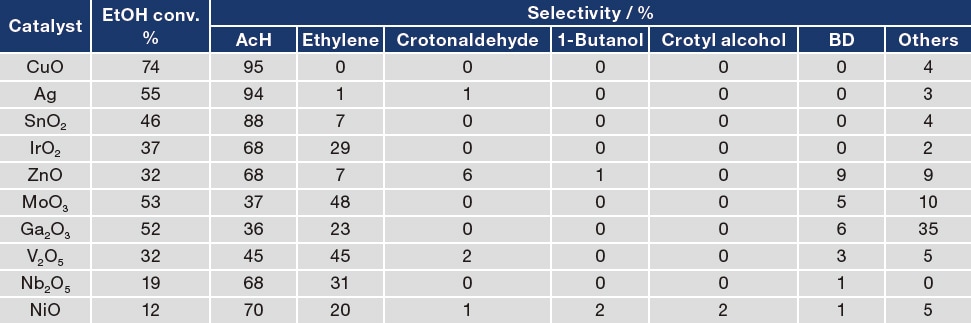
EtOH: 0.03 mL/min; N2: 10 mL/min; Reaction temperature: 623 K; Support: SiO2; Loading amount of metal oxides: 5 wt%.
We next screened for catalysts that effectively catalyze the second-stage reaction converting AcH to BD. Again we tested a total of 200 candidate catalysts. Table 2 lists results for a representative subset of 10 catalyst candidates. We chose an EtOH/AcH ratio of 50/50 and measured catalytic activity at 623 K. Four catalyst candidates—SiO2-supported HfO2, ZrO2, Sc2O3, and Nb2O5—exhibited high rates of EtOH conversion and high BD selectivity3). HfO2 and ZrO2 were previously known to be excellent ETB catalysts, but our findings for Sc2O3—namely, that Sc2O3 is also an effective catalyst component for the portion of the ETB reaction following EtOH/AcH—is a new discovery. Next, for each of the four catalysts exhibiting high activity, we attempted to optimize various conditions and parameters to maximize yield. The results of this process indicated that the optimal catalyst is HfO2 with a loading amount of 7 wt%, for which we obtained a maximum yield of 63% at a reaction temperature of 673 K and an EtOH/AcH molar ratio of 1.5.
We next developed a model, based on the data described above, for predicting the BD yield for the EtOH/AcH reactions. Our model applies a random-forest approach with a learning-data number of 180 and a test-data number of 20 (Figure 3). From this we learned that the most important factor affecting the BD yield is the charge of metallic electrons in metal oxides, while the BD yield is also highly sensitive to the distances between metal atoms and oxygen atoms. This finding is intuitively reasonable from the perspective of reaction mechanisms, considering that metallic regions are positively charged and function as Lewis acids, while oxygen regions are negatively charged and function as Lewis bases.
Table 2 EtOH/AcH reaction activity for a subset of catalyst candidates prepared by a high-throughput catalyst synthesizer.
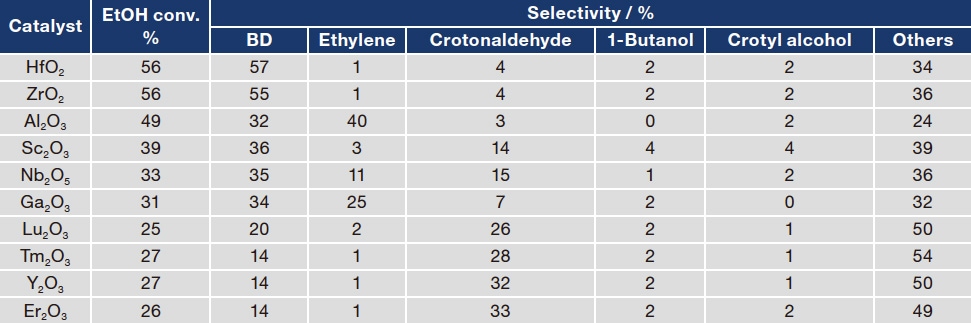
EtOH/AcH: 50/50, 0.03 mL/min; N2: 10 mL/min; Reaction temperature: 623 K; Support: SiO2; Loading amount of metal oxides: 5 wt%
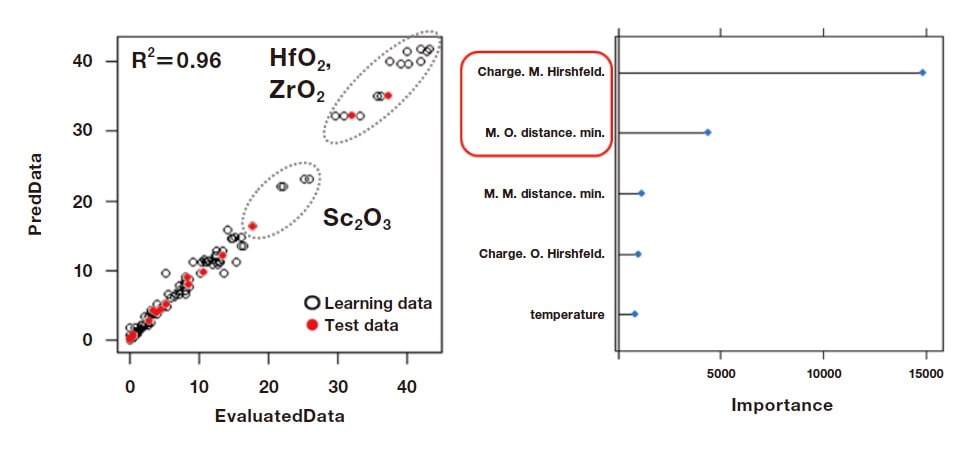
Fig. 3 Predicting BD yield from EtOH/AcH conversion reactions catalyzed by metal-oxide catalysts.
Algorithm: Random Forest, Learning data: 180, Descriptors: 5, Test data: 20
Based on the high-throughput screening evaluations discussed above and the collection of approximately 200 measured datasets they produced, together with a statistical analysis using data from theoretical calculations, we conclude that an Ag/SiO2 catalyst was most effective for the first-stage reaction, while an HfO2/SiO2 catalyst was most effective for the second-stage reaction. As a next step, we designed a tandem flow reactor and used it to assess ETB activity in the two-stage reaction for the combination of these catalysts; results are listed in Table 3. For the one-stage reaction, we observe low BD selectivity (48%) for Ag/SiO2 + HfO2/SiO2 physical mixture and Ag-HfO2/SiO2 catalysts. On the other hand, SiO2-supported ZnO and ZrO2 catalysts gave low EtOH conversion rates (67%). For the twostage reactions, by contrast, setting the reaction temperatures to 603 K and 673 K—the optimal temperatures for the respective reactions—yields an EtOH conversion rate of 99% and a BD selectivity of 63%3).
Even more interesting is the following observation: In the one-stage reaction, Ag/SiO2 + HfO2/SiO2 physical mixture, Ag-HfO2/SiO2, and ZnO-ZrO2/SiO2 catalysts exhibited a significant decline in catalytic activity as the reaction proceeds. In contrast, for the two-stage reactions catalyzed by Ag/SiO2 and HfO2/SiO2, the BD yield remained essentially stable, indicating that the mechanism responsible for degrading catalytic activity as the reaction proceeds was suppressed in this case. Our findings also demonstrate that the two-stage reaction process is effective from the standpoint of catalyst lifetimes. At present, in addition to continuing to improve these catalysts, we are using high-throughput instruments to gather various types of engineering data and exploiting process informatics to conduct larger-scale tests of our methodology in preparation for deploying it in practice.
Table 3 ETB reaction results for various combinations of catalysts.
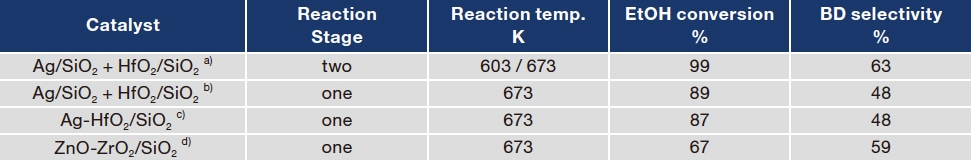
EtOH feed rate: 0.03 mL/min; N2: 10 mL/min.
a) Ag and HfO2 loadings: 7 wt% b) Physical mixture of Ag/SiO2 and HfO2/SiO2, Ag and HfO2 loadings: 7 wt%
c) Ag and HfO2 loadings: 7 wt% d) ZnO and ZrO2 loadings: 5 wt%
The challenge of reducing the quantity of Pt used as a positive-electrode catalyst in solid-polymer fuel cells has emerged as a crucial imperative. One possible strategy is the use of core-shell catalyst particles—in which Pt is present only at the outermost surface layer—and the development of core-shell catalysts has become an active area of research. However, existing methods for synthesizing core-shell catalysts rely on batch processes, whose low productivity and other disadvantages pose major hurdles for practical deployment. This motivated us to investigate the possibility of developing flow synthesis methods for core-shell catalysts, with the goal of establishing a synthesis technique offering both high productivity and sophisticated control over catalyst structures. Achieving flow synthesis requires optimizing various conditions affecting the formation of Pt shells to develop a flow technique for preparing Pd@Pt catalysts with Pt-shell structures featuring Pt monatomic layers. To this end, we developed a high-throughput flow-synthesis system for synthesizing core-shell particles (Figure 4), then rapidly optimized various process parameters and conditions, including metal precursors, reactants, types of additives, contact efficiency, and residence times. This synthesis system uses a plunger pump to supply various metal precursors and reducing agents from an autosampler to a reactor. At the reactor, precursor of Pd—the core metal— is first reduced by the reducing agent to form Pd-core particles. Then, in a second step, Pt precursor are reduced by mixing with the reducing agent, and Pd@Pt core-shell particles are formed from Pd core particles and reduced Pt metal. After Pd@Pt particles form, they are fed into a carbon slurry and supported on an activated carbon. Finally, fraction collector is used to transfer the activated-carbon-supported Pd@Pt core-shell catalysts to vials for storage. The use of this system as outlined above allows continuous automated synthesis of around 20 varieties of Pd@Pt catalysts per day.

Fig. 4 High-throughput flow-synthesis system for Pd@Pt catalysts.
As a first experiment, we used H2PtCl6 as a Pt precursor and investigated how the Pt shell structure and catalytic activity are influenced by the type of reducing agent. Pt–Pt and Pt–Pd coordination numbers for synthesized Pd@Pt catalysts were determined by measuring XAFS spectra and fitting the data to appropriate functional forms. We used oxygen reduction reaction (ORR) activity as a measure of catalytic activity. When 2-MePy•BH3 was used as reducing agent, we estimated values of 6.8 and 2.5 for the Pt–Pt and Pt–Pd coordination numbers, respectively. Because these values are close to the exact theoretical values (6 and 3 for Pt–Pt and Pt–Pd, respectively) expected for the case of a Pt shell consisting of only a monatomic layer, we considered the formation of controlled Pd@Pt core-shell particles with a nearly single layer Pt shell. However, for these Pd@Pt catalysts, the observed values of electrochemical active surface area (ECSA), ORR activity per unit Pt mass (MA), and ORR activity per unit Pt surface area (SA) are only around 50% of those measured for Pd@Pt catalysts synthesized by the traditional method of copper–underpotential deposition (Cu–UPD). The low values observed for Pt surface area and ORR activity—despite the existence of a monatomic-layer Pt shell—are due to the agglomeration of Pd@Pt particles, as shown in Figure 5.

Fig. 5 DF-STEM image of Pd@Pt catalyst formed with H2PtCl6 as Pt precursor and 2-MePy•BH3 as reducing agent.
Examples in which capping agents are used to disperse Pd@Pt particles for supporting on an activated carbon have been previously reported6). Pyridine-based compounds that coordinate with Pt in nitrogen have also been used as capping agents7-9), and it is conceivable that 2-MePy, produced by the reaction of Pt precursors with 2-MePy•BH3 reducing agent, may function as a capping agent. However, when a 2.1-equivalent quantity of 2-MePy•BH3 was added as a reducing agent for Pt precursors, we were entirely unable to suppress the agglomeration of Pd@Pt particles. Therefore, we increased the amount of additive and investigated its impact on the Pd@Pt structure. Table 4 lists measured ORR activities, and Pt–Pt and Pt–Pd coordination numbers determined by data-fitting analysis of EXAFS spectra, for various quantities of 2-MePy•BH3 additive. When the quantity of 2-MePy•BH3 additive is below 6.4-equivalent, the Pt–Pt and Pt–Pd coordination numbers are close to theoretical values for the monoatomic layer Pt shell structure, indicating that the uniform monatomic-layer structure of the Pd@Pt core-shell particle is preserved. ECSA and MA increase as the quantity of reducing agent increases, with MA peaking at a maximum value of 522 for an additive quantity of 6.4-equivalent (the catalyst labeled "Cat. 3" in Table 4). This value is comparable to values observed for a Pd@Pt catalyst prepared by the Cu–UPD method10). However, further increasing the amount of 2-MePy•BH3 causes MA to decrease. Figure 6A shows a TEM image of the activity-maximizing catalyst ("Cat. 3"). We observed that the dispersion of Pd@Pt particles is significantly improved for Cat. 3 compared to the case of Cat. 1. This finding is consistent with ECSA results and indicates that 2-MePy derived from 2-MePy•BH3 functions as a dispersing agent. EDS mapping analysis of a single Pd@Pt particle (Figure 6B) reveals a spherical Pd particle (green) with Pt (red) uniformly distributed over its surface, confirming the Pd core / Pt shell structure.
Next, Figure 6C shows the results of electron energy-loss spectroscopy (EELS) line analysis to observe the finegrained structure of the Pt shell. From the EELS data we estimate that the thickness of the Pt shell is approximately 0.25 nm. As the atomic diameter of Pt is 0.28 nm, we clarify that the Pd@Pt core-shell particles we synthesized have Pt shells consisting of a single monolayer. From these findings, we conclude that presence of 2-MePy serves two purposes: (1) forming the monatomic-layer structure of the Pt shell, and (2) suppressing the tendency of Pd@Pt particles to agglomerate. More specifically, Pt atoms deposited at the surface of the Pd core particle are coordinated with 2-MePy, and this 2-MePy obstructs Pt crystal growth, resulting in the formation of a Pd@Pt particle with a monatomiclayer Pt shell. This also shows that optimizing the quantity of 2-MePy can reduce the tendency of Pd@Pt particles to agglomerate.
Table 4 Structure and catalytic properties of Pd@Pt catalysts synthesized with various quantities of 2-MePy•BH3 additive.
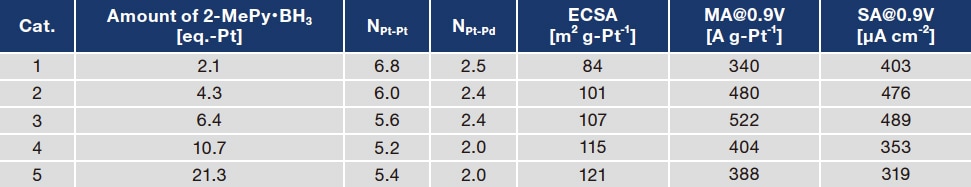
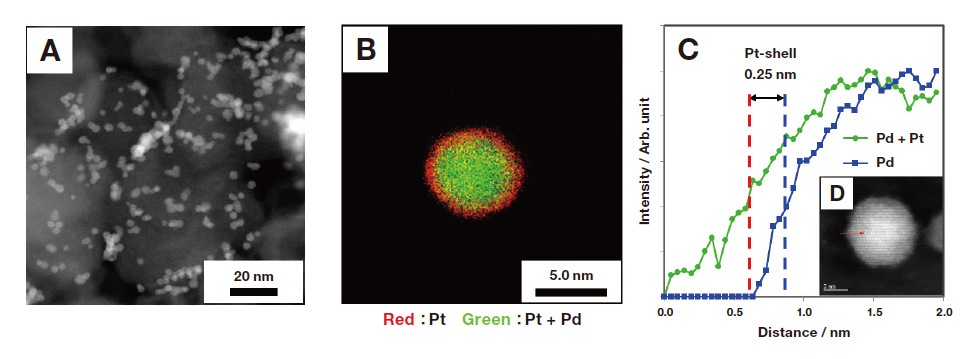
Fig. 6 TEM image (A), EDS mapping image (B), and EELS line analysis (C,D) for a Pd@Pt catalyst synthesized with H2PtCl6 as Pt precursor and 2-MePy•BH3 (6.4 eq.-Pt) as reducing agent. (This is the catalyst labeled "Cat. 3" in Table 4).
In this paper we investigated a data-driven approach to catalyst development that combines informatics techniques with high-throughput laboratory instruments capable of rapidly preparing and evaluating the performance of catalysts to accumulate high-quality experimental data. Our results demonstrated that this data-driven strategy allows the development of high-performance catalysts, and of new synthesis methods, in short periods of time. At present we are actively improving our collection of high-throughput laboratory systems and accumulating experimental data on a variety of catalytic reactions using high-throughput catalyst preparation system, high-throughput catalytic activity evaluation system, and high-throughput instruments for various other types of analysis. We have also set up a platform for catalyst development, including a full range of instruments, at the AIST Tsukuba Center; we hope this new research facility will provide many researchers with the opportunity to use high-throughput techniques and discover the new world of possibilities made available by data-driven catalyst development.
Acknowledgements
We are grateful to have received support for this research from NEDO.
References
See more
