
Kiyoharu Nakatani
Ph.D (Engineering)
Professor
Institute of Pure and Applied Sciences
University of Tsukuba
Porous particles offer high specific surface area and unique porespace properties that make them useful for a variety of applications, including adsorbents, catalysts, and separation techniques such as chromatography and solid-state extraction. Our research group studies aspects of analytical chemistry relevant for separation and refinement, with a particular focus on mass transfer in porous particle systems used for chromatography and similar purposes. In systems involving porous particles in solutions, mass-transfer mechanisms carry solutes back and forth across the boundaries separating particle surfaces from the solution phase outside the particles; when this happens, solutes are drawn into pores on particle surfaces and are repeatedly adsorbed and desorbed while diffusing through the interiors of particles. We believe that phenomena of this sort are best analyzed by separating them into aggregates of elemental processes, and to facilitate such a separation we make spectroscopic measurements of individual micron-scale spherical particles under microscopes. Mass transport from a bulk solution across the surface of a micron-scale particle, even in the case of diffusion only, proceeds efficiently via steady-state diffusion, allowing easy analysis of processes within the particle. In this study we use single-particle measurements to analyze intra-pore diffusion in silica gel systems.
Figure 1 shows a schematic diagram of our single-particle microspectrophotometry setup. In this method, a single microparticle is injected under a microscope via microcapillary manipulation, and micro-absorption or (confocal) micro-fluorescence measurements—with probe light focused to micron-scale spot sizes—are used to measure distributions within the microparticle or emission processes from the microparticle. Particles of larger sizes tend to remain at rest on the cell floor, but smaller particles may exhibit Brownian motion, in which case we use laser trapping to hold the particle in place. Taking the instant of particle injection to be the origin of the time axis, we observe the temporal evolution of absorption or fluorescence intensity and analyze intra-particle diffusion processes on the basis of kinetic theory.
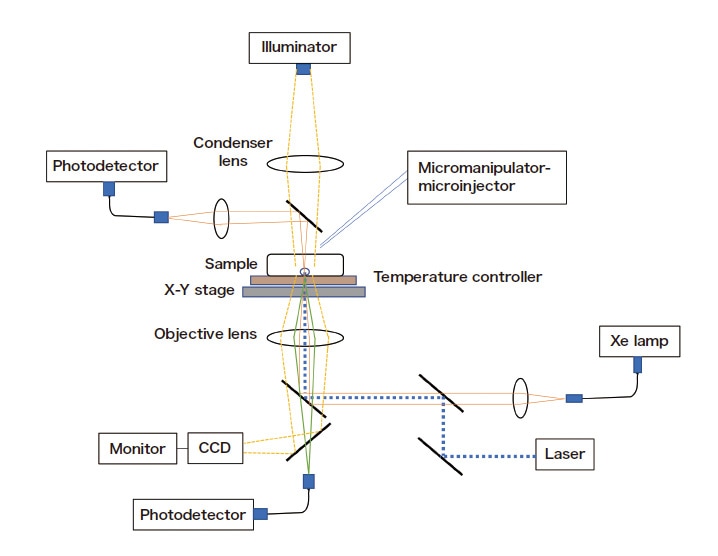
Fig. 1 Schematic diagram of experimental setup for single-particle microspectrophotometry.
Due to the dissociation of silanol groups, silica gel contains negatively-charged adsorption sites at which cationic solutes are adsorbed. Figure 2 shows the results of confocal micro-fluorescence measurements of a single silica-gel particle, with a pore diameter of 6.5 nm, added to an aqueous rhodamine 6G solution. Because the particle lies on the cell floor, the fluorescence profile in the depth direction along its central axis allows us to observe efficient diffusion into the center of the particle from its upper surface (the upper and lower particle surfaces are at depths of 30 μm and –30 μm respectively). The intra-particle diffusion coefficient Do may be determined from simulations of diffusion inside and outside the particle (taking into account the suppression of diffusion at the cell floor)1). To analyze diffusion in porous media, we use a model that takes into account three distinct processes (Figure 3): rapid adsorption and desorption at pore walls and between the solution and the pore interior, diffusion through the solution within pores (pore diffusion), and diffusion along pore walls (surface diffusion). In this model, the diffusion coefficient is given by
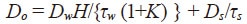
where Dw is the diffusion coefficient in the aqueous phase, H is a pore-wall parameter, K is the distribution coefficient (K>>1), and τw and τs denote curvature rates. Thus, plotting Do versus 1/(1+K) should yield a straight line whose slope and offset are DwH/τw related to pore diffusion and Ds/τs related to surface diffusion, respectively.
Measurement of the K dependence of Do indicates that, for a small molecule such as rhodamine 6G, pore diffusion is the dominant process, with surface diffusion making almost no contribution. We also note that, by coating the pore walls of silica-gel particles with styrene-divinylbenzene copolymer and embedding extraction agents inside the particles, we obtain carriers for extraction chromatography to separate metal ions from highly radioactive waste liquids. For particles ≤30 μm in size with poresizes of a few hundred nanometers and extraction agents for which the reactions required to form complexes with ions such as Eu(III) or Sm(III) proceed rapidly, pore diffusion is known to be the rate-limiting step in the extraction process2).
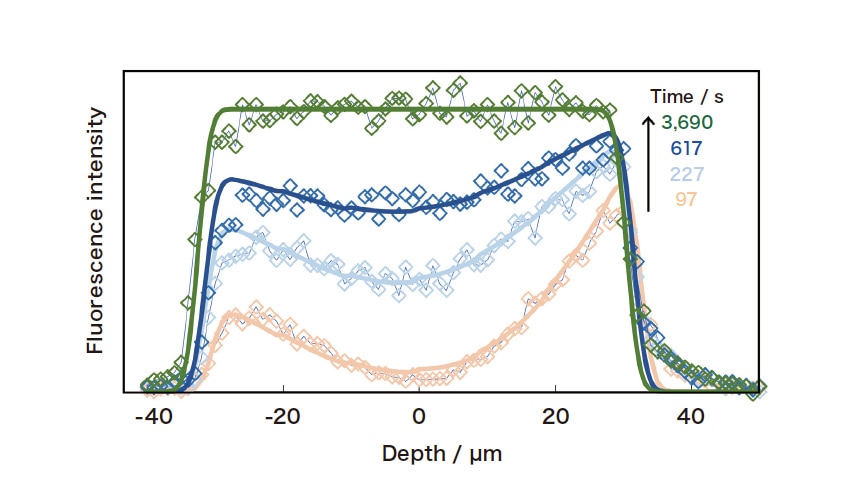
Fig. 2 Confocal micro-fluorescence measurements of rhodamine 6G inside a single silica-gel particle. Solid curves indicate the results of diffusion simulations (with Do = 6 × 10-9 cm2/s).
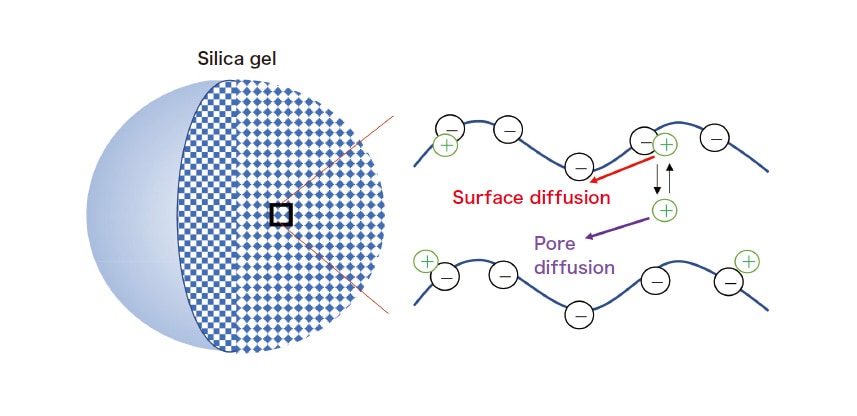
Fig. 3 Diffusion processes in porous silica gel.
The nature of intra-particle diffusion in silica-gel systems changes significantly if we replace small molecules with macromolecules. Measurement of the distribution speed for spherical fluorescent macromolecules (with an approximate diameter of ≤4.4 nm, prepared by replacing a heme of myoglobin with zinc porphyrin) through silica gel with 15-nm pores, indicates that surface diffusion is the dominant process in this case, with pore diffusion making only a minor contribution3).
Next, adding a single particle of ODS silica gel—a reverse-phase chromatography carrier—to a solution and measuring its distribution process reveals that, although the fluorescence intensity from the particle varies with time, no concentration gradient is observed inside the particle. This is due to the extreme rapidity of intra-pore diffusion; for this measurement technique, diffusion outside the particle is the rate-limiting step. Instead, to measure intra-particle diffusion in systems such as this we irradiate the particle interior—which is in equilibrium with the distribution— with high-intensity laser light to induce a non-equilibrium state known as photobleaching, and then use confocal micro-fluorescence measurements to observe the relaxation process as the system re-equilibrates. By simulating the concentration distribution within the particle, we determine intra-pore diffusion coefficients for coumarin 101 and other species in ODS silica gel (Figure 4); the results indicate that surface diffusion, in which the solutes move through the ODS phase on pore walls, makes a significant contribution4).
In future work, we plan to investigate how intra-pore mass transfer in porous particles is affected by factors such as molecule/poresize ratios, molecular shapes, and electric double-layer effects from pore walls; we are hopeful that these investigations will shed light on chromatography separation mechanisms and other phenomena of interest.
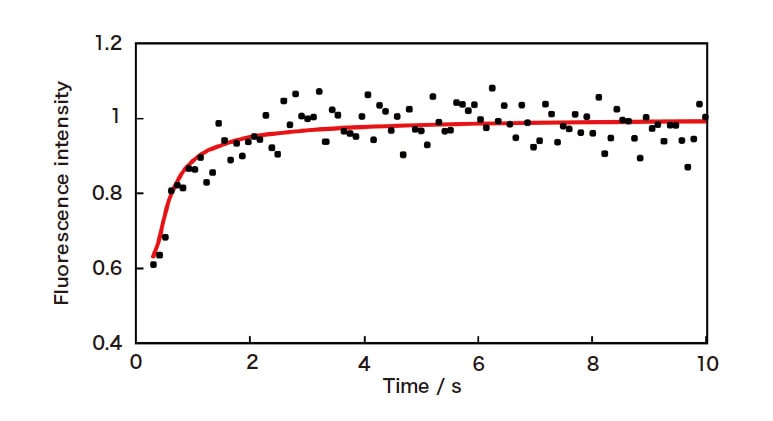
Fig. 4 Confocal micro-fluorescence measurement of relaxation from a photobleached state for coumarin 101 inside a single particle of ODS silica gel (particle size 34 μm). The solid curve indicates the results of diffusion simulations (with Do = 2 × 10-8 cm2/s).
References
