
Masami Shibukawa
PhD in Science
Professor Emeritus
Graduate School of Science and Engineering
Saitama University
Methods for separating chemical substances may be broadly classified into two categories: (1) methods that exploit a partition equilibrium between two phases or an adsorption equilibrium between a phase and an interface, and (2) methods that achieve separation by applying an externally-generated physical force (sometimes known as an outer field) to a single phase. Distillation, precipitation, liquid-liquid extraction, and the various forms of chromatography belong to the former category, while electrophoresis, ultracentrifugation, and field-flow fractionation belong to the latter category. At present, most of the methods commonly used to separate substances—particularly for preparative-scale separations—belong to the former category.
In methods belonging to the former category, the system in which separation takes place consists of two components: the phase and the interface. Consequently, a clear understanding of the structures and physical properties of the phase and the interface, in particular, interaction with solute molecules, is an essential prerequisite for understanding mechanisms of separation and for predicting the separation performance (as measured by resolution, separation factor, or other benchmarks) achievable by a given choice of separation method. But how, precisely, are the phase and the interface to be defined? According to the Standard Dictionary of Chemical Terms (2nd Edition, 2005) compiled by the Chemical Society of Japan (CSJ), a phase is “a part of a physical system, of uniform physical and chemical properties, separated from other parts of the system by clearly-defined interfaces”, while an interface is “the boundary surface formed between two distinct phases—gas, liquid, or solid—in contact with each other”.
One immediate question arises at this point: precisely how many molecules must aggregate before we can state that a “phase” has formed? For methods such as distillation or liquid-liquid extraction, the “phases” in question are typically visible—or, at least, are large enough to be visible—to the naked eye, so questions regarding their sizes generally do not arise; however, the same cannot be said for chromatography methods. For example, in liquid chromatography(LC) which uses columns packed with solid materials, the separation mechanism is described in terms of molecular or ionic partitions between the mobile phase and the stationary phase; in many cases, however, the stationary phase, and sometimes the mobile phase as well, exist only as figments of the imagination, with the respective sizes of these phases rarely quantified with high precision.
The term “phase” is also defined in the Kagaku Dai-Jiten (Encyclopedia of Chemistry) (Kagaku-Dojin, 1989), which has this to say regarding its size: “When all parts of a g iven thermodynamic system exhibit identical physical a nd chemical properties, the full system is referred to as a single phase. The apparent sizes of phases are of no relevance, but phases cannot be distinguished at the sub-molecular level.” In other words, only aggregates of molecules or ions can exhibit the properties of gases, liquids, or solids required to constitute a “phase;” an individual molecule cannot be considered a phase. This reasoning suffices to establish that the number of molecules required to constitute a phase is greater than 1, but nonetheless leaves our question largely unanswered; for example, can we truly state definitively that an aggregate of 2 or 3 molecules could constitute a phase? It seems reasonable to suppose that no such definitive statement can be made. I personally remember participating in a seminar for young researchers, organized by the CSJ more than 35 years ago, at which I addressed this question to the lecturer (whom I recall to have been an expert in polymer physics). He helpfully explained that, although no precise answer can be given, some foreign researchers would argue that an aggregate of N molecules could be properly called a “phase” whenever N is large enough that Stirling's formula (an approximation formula familiar to students of mathematical analysis and statistical mechanics) holds. Playing around with the formula, one finds that the approximation is reasonably well established at a slightly larger number of molecules than 10.; of course, new questions arise when we ask to what extent (i.e. to how many digits of accuracy) Stirling's approximation holds. Ultimately, there is no unambiguous physicochemical basis for resolving this question, and we are forced to conclude that there exists no one definition that applies to all systems.
Returning to the example of LC methods, here again it is easy to think of the mobile phase as a “phase”—after all, before the solution (the mobile phase) is introduced to the column we can see it with our own eyes, sitting right there in a solvent reservoir! Thus, even after the mobile phase has been introduced into the column it is reasonable to think of this phase as retaining the same state in which it previously existed in the reservoir—at least, this is true in the regions between the particles of the packing material, or, for monolithic columns, in macropores. On the other hand, it is less easy to picture the structure and nature of the stationary phase that forms in the column; in particular, the material with which the column is packed (the packing material) does not simply become the stationary phase, with no change in form, after coming into contact with the mobile phase liquid inside the column. The key factors to consider are the size and structure of the interfaces that form when the packing material comes into contact with the mobile phase liquid. As noted above, “interfaces” are defined as boundary surfaces between phases, but interpreting this phrase literally would imply that interfaces were strictly 2-dimensional entities, with zero thickness. The Kagaku Dai-Jiten has this to say: “Considering that the surfaces or interfaces of a substance exhibit properties different from those of the bulk substance, they (together with any aggregates of molecules adsorbed on them) may be thought of as 2-dimensional-like phases, distinguishing them from ordinary phases.”
This notion—that a phase has nonzero thickness, while an interface does not—creates a distinction between the phenomena of partition and adsorption. Partition is a phenomenon in which solute molecules move between two phases to yield an end state in which, within each phase, solute molecules are completely surrounded by the molecules comprising that phase. Adsorption, by contrast, is described as a phenomenon in which molecules from one phase aggregate at the interface between phases (positive adsorption) or are repelled from that interface (negative adsorption). Thus, for example, in a positive-adsorption process in which solute molecules from a liquid phase adsorb at a liquid solid interface (i.e. on the surface of the solid phase), we can think of the solute molecules as replacing solvent molecules that were originally present at the interface. However, this simplified picture cannot explain separation phenomena in systems featuring extremely large interfaces between phases, as is true not only for LC but also for some liquid-liquid extraction techniques (such as methods using microdroplets).
By way of analogy, an exercise that proves instructive at this point is to consider the different behaviors of hydrophilic and hydrophobic solids when brought into contact with water. Methods exploiting solid-liquid or liquid liquid interfaces are among the most widely-used techniques for separating chemical substances, particularly in aqueous solutions; examples of such methods include LC and solid-phase extraction (SPE), both of which use the interface as one of the primary drivers of separation. Many varieties of substances can be used as separating materials to form an interface with an aqueous phase; when these materials come into contact with water, their surfaces transition to various states that are not uniquely determined. For example, when hydrophilic polymers such as dextran, polyacrylamide, or polyethylene glycol are used as base substances or surface coatings for column packing materials or solid-phase extraction agents, some or all of them mixed with water to form a liquid phase that functions as a separating medium1-3). In such cases, the hydrophilic polymers are either crosslinked or fixed to the base materials by covalent bonds, preventing them from dissolving into the aqueous phase and allowing them to behave as the stationary liquid phase. We note, however, that the liquid phase thus formed will not necessarily be a simple random mixture with water molecules, but may, in some cases, assume certain specific structures4). In such cases, the thickness of the polymer-solution phase that serves as the stationary phase may—for the case of a polymerized macromolecular gel—expand in water to the size of the gel itself (which, for LC column packing materials, ranges from a few micrometers to a few tens of micrometers), yielding a process more closely resembling partition than adsorption.
On the other hand, bringing water into contact with a hydrophobic surface results in the formation of round water droplets. This is a consequence of strong mutual attraction between water molecules due to hydrogen bonds, but also means that water molecules tend to avoid contact with hydrophobic surfaces. Thus, forcibly bringing water into contact with hydrophobic surfaces results in the formation of well-defined interface separating the water phase from its counterpart phase. A system of this sort may be created by forcing water at high pressure through a column packed with C18-bonded silica gel (a silica gel whose surface has been chemically modified with octadecylsilyl groups, making it the most general-purpose choice for reversed-phase LC). (Note that pure water is rarely used as the mobile phase for typical reversed-phase LC methods, which instead use aqueous solutions to which methanol or acetonitrile are added.) However, the physical properties and structure of water in the vicinity of hydrophobic material surfaces have been shown by many studies to differ significantly from those of bulk water. In particular, water near a hydrophobic surface assumes an irregular structure: the orientations of, and the hydrogen bonding between, water molecules touching the surface differ from those of bulk water molecules5,6). Thus, we can say that, although the interface separating water from a hydrophobic material is in fact 2-dimensional, the liquid phase formed by water near the interface differs from the liquid phase that prevails at points far from the interface. (Note that the surfaces of C18-bonded silica gels are typically not smooth, but exhibit surface roughness.) The existence of the interfacial water layer—a region of nonzero thickness, extending a finite length in the direction perpendicular to the interface, over which the structure of liquid-phase water is modified—has important consequences for the solute molecules that are the target of the separation procedure: Solute molecules that enter this interfacial layer may be considered, in a macroscopic sense, to have adsorbed on the interface, even if the molecules do not actually reach the interface.
We used ions as a probe to measure the thickness of the water layer at an interface between water and C18-bonded silica, obtaining a value of approximately 1.3 nm5). This length, which corresponds to the thickness of 4 to 5 layers of water molecules, indicates the distance at which ions and molecules can sense the presence of the interfacial water or detect the presence of the hydrophobic surface. A schematic diagram illustrating the vicinity of an interface between water and C18-bonded silica is shown in Figure 1; this figure indicates the thickness of the interfacial water region and the distributions of methanol, nitromethane, and benzene7-10). Interestingly, we see that, whereas methanol is present primarily in the bulk water and in the interfacial water layer, benzene is present in the interfacial water layer and in the alkyl-chain layer. On the other hand, nitromethane is not only present in bulk water and in the interfacial water layer, but is also distributed over the end-capped silica surface. Meanwhile, strongly hydrated inorganic ions are prevented from entering the interfacial water layer, resulting in the phenomenon known as negative adsorption5). These findings are in good agreement with recently reported results of molecular dynamics simulations11-13).
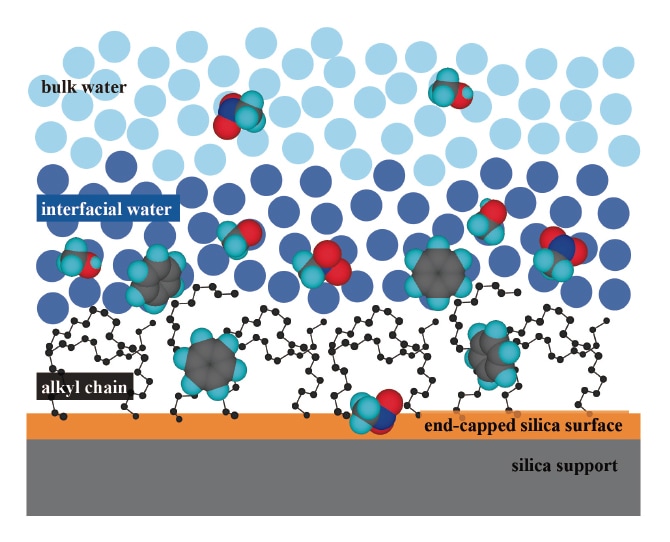
Fig. 1 Schematic illustration of an interface between water and C18-bonded silica, indicating the interfacial water layer and the partitioning behavior of solutes.
As this discussion demonstrates, the existence of an interface between two phases modifies the structures of the phases near the interface; the thickness of the region over which this modification takes place depends on the type of solvent used—and, for mixed solvents, on the composition—but is at least on the order of 1 to several nanometers. We have used molecules and ions as probes to measure these interfaces, and there have been many other reports of studies using various approaches—including sum-frequency generation spectroscopy and other interface-selective spectroscopy methods, atomic force microscopy, and molecular dynamics calculations— to clarify the properties of various types of interfaces. The progress of this research is spurring an evolution in modern thinking beyond the conventional picture in which phases and interfaces, and the associated phenomena of partition and adsorption, were treated as unequivocally distinct entities.
References
